So you have some data, perhaps a lot of data, but you’re not quite sure what to do with it… where do you start? Is it interesting data? Would it tell a good story, or just be a confusing mess? This is exactly our problem at work. We have a lot of our own data, and we have access to a stupid amount of publically available data. But no one is quite sure what to do with it. So I have begun playing with a nice little set of gene expression data. Here’s how it goes…

The question
A good mystery always has a great plot and a compelling question. In this case, we want to know if there are any interesting groups of genes in our expression data? Can we find groups of genes which are similar to each other? Can we perhaps find groups of genes which are unlike other genes?
These are really interesting questions, and might help shed light on biological function or biological pathways.
The data
OK – the data, the most important part. We’ve downloaded a good-sized (5 GB) set of gene expression data from the GTEx Portal. The dataset includes RNA read counts for 56318 genes (the rows) across 8555 tissues (the columns). To help build context to this dataset, we have also downloaded a couple of additional data sets which will help us map the expression data to common tissue types and phenotypes.
The challenge
I’m excited about this dataset. It’s not huge, but it is big enough that the size of it is a fun problem by itself. I’ve been looking for an excuse to play around with R’s data.table package for a while now, and this seems like a great opportunity to give it a whirl. So, from a selfish perspective, this analyses has been about playing around with data.table. From a less-selfish perspective, this analysis is about finding simple ways that we can visualise gene expression data and add layers of additional information / context. The methods need to be intuitive, simple and quick so that I can wrap them up into a nice-and-easy interactive app for everyone else.
The plan
I think there is an unwritten law in genomics – all analyses and papers must have a heatmap! So I will begin with a heatmap. They are a really good way of visualising high-dimensional data. But extracting anything meaningful out of them can be a hard slog. So I will also look at ways to layer extra information onto the heatmap.
Although groups of similar genes should be “immediately obvious” from the heatmap, identifying clusters in a heatmap isn’t always that easy. So we will use heirarchical clustering (sensible right, seeing as this is what a heatmap uses anyway?) to define the clusters and add these as a coloured layer to the heatmap.
And just to prove we aren’t crazy, we will try using factor analysis to see if we can see the same gene clusters in the factor loadings.
The base heatmap
The most basic heatmap is at the top of this post. No clustering, no reordering of the rows – just a direct plot of the genes (columns) across all the tissue samples (rows). R’s base heatmap() function will order the rows and columns for us. Optionally, you can also choose to display the row and column dendrograms. Here is the resulting heatmap:

This is a definite improvement. We can begin to see some gene clusters. In particular, there is an obvious cluster of genes to the far left of the heatmap. The column dendrogram suggests that there are a couple of potential clusters in the right-hand third of the plot. I am not very good at deciphering these things, but even I can see two strong clusters: the ‘red’ group in the center of the heatmap and the group or 3 or 4 genes just to right of these (TFP1, TPPP, ABCG2, SFXN5).
Heirarchical clustering
If I am honest though, I find heatmaps quite overwhelming and I would prefer to look at individual dendrograms. If we cluster the genes in R, then we get the following:
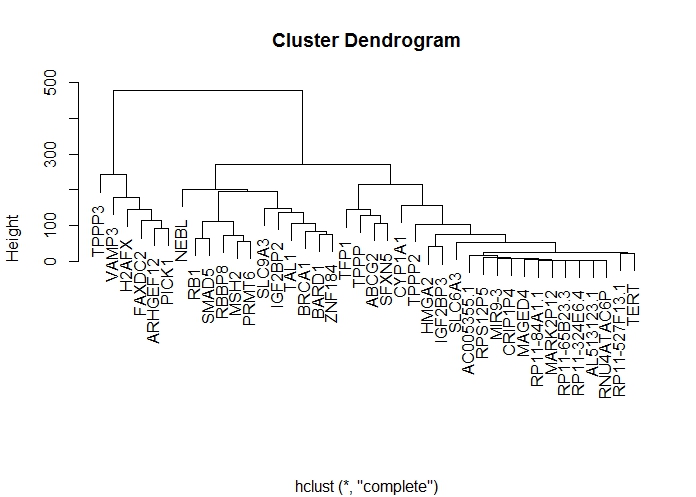
TIP: to identify clusters of genes using hclust(), your data matrix should have genes in the rows and expression data in the columns (i.e. each observation (row) is a unique gene, described by a series of variables, in this case, expression data).
I definitely prefer this dendrogram over the full heatmap, it just makes it easier to see the similarities / clusters of genes. So what I am going to do is cut this tree just above Height = 100, which will give us approx. 8 clusters. Then, I will replot the heatmap but also include a set of colours that mark the cluster boundaries:

This is great – the clusters of similar genes are now much more pronounced. In particular, TPPP3 and VAMP3 really stand out at the far left, and we can definitely see the group (TFP1, TPPP, ABCG2 and SFNX2). Also interesting is NEBL which is all by itself in the middle there.
Factor Analysis
The clusters above are really compelling. And I have pretty much convinced myself that they must be real. But if they are real, then we should be able to reproduce these clusters (or something very similar) using another clustering technique. In fact, we should be able to go even further and identify similar groups of genes in a non-clustering technique such as factor analysis.
Clustering and factor analysis are certainly not the same thing. Clustering takes objects (observations, cases, rows) and tries to find groups of similar groups based on their features (columns). Factor analysis is different, it takes the features (columns) and tries to find combinations of these columns which describe the object (observations, cases, rows -whatever you want to call them). It is kind of the same, but it really isn’t at all similar.
However, if we really have found meaningful clusters of genes above, then we should be able to take the tissue samples (objects) and find combinations of genes (features) which can differentiate between the tissues. So I set this as my challenge, transposed the dataset so that I had tissue samples down the rows and genes along the columns. And here’s what I got:

OK – I admit – the plot above is not easy to interpret. The x-axis is a plot of all the genes, ordered by there cluster ID (the same cluster ID that we got from heirarchical clustering). The colours also represent the cluster IDs (just the same way that the coloured bar above the heatmap did). The y-axis represents how important (+1 is ver important, -1 is also very important) to each factor. What we can see is that each factor is clearly dominated by one or two groups. And, if we plot this slightly differently and highlight the gene names we get something a little more obvious:

We still have to jump back a forward between the heatmap and this plot. But if you do, you will notice that each factor is dominated by a very well-defined region of the heatmap. Specifically:
- Z1 is a combination of genes which are all to the far right of the heatmap
- Z2 is dominated by SFXN5, TPPP, ABCG2 and TFP1 which are all part of the same cluster on the heatmap
- Z3 is dominated by genes which all fall in the center (the ‘red’ zone) of the heatmap
- and so on, and so on…
What does this mean? Not much really, except that we have managed to take some tentative gene clusters from the heat map and reproduce these using an entirely different method. This is reasonably strong evidence that the clusters are robust. Are they biologically meaningful? That I’m not so sure about. There are a few issues with the data ( for example a massive imbalance in the number of samples per tissue), and I really don’t know enough about these genes to say. But hey – that’s not my job. My job is to develop tools to help the people who do know about this tuff to explore their data in meaningful ways.
